LOT
Citation
Condition of Use
- LOT can be used and distributed free of charge under the following terms and conditions.
- Any research using this program or the methods or ideas behind it should acknowledge the use of LOT, and cite the references in the Citation section.
This software is provided by the collaborative center for statistics in science at Yale University as is, and any express or implied warranties, including, but not limited to, the implied warranties of merchantability and fitness for a particular purpose are disclaimed. In no event shall the collaborative center for statistics in science at Yale University be liable for any direct, indirect, incidental, special, exemplary, or consequential damages (including, but not limited to, procurement of substitute goods or services; loss of use, data, or profits; or business interruption) however caused and on any theory of liability, whether in contract, strict liability, or tort (including negligence or otherwise) arising in any way out of the use of this software, even if advised of the possibility of such damage.
Versions
- LOT 1.2: April 24th, 2008
- LOT 1.1: February 18th, 2008
Methodology
Four Main Steps of LOT
I. Data Input
This part is modified from the Genehunter program to accommodate the ordinal trait and to add the graphic user interface (GUI). See Input file formats for the details.
II. Inference of Inheritance Vectors
This part is taken from the Genehunter program. The method is described in (Kruglyak et al., 1996). LOT infers the inheritance pattern of a pedigree by means of inheritance vectors, v, which is irrelevant to the type (continuous or categorical) of the trait. The inheritance pattern at a marker location is completely described by an inheritance vector  whose elements describe the outcomes of the paternal and maternal meioses transmitted to the n offspring in a pedigree. Specifically,
whose elements describe the outcomes of the paternal and maternal meioses transmitted to the n offspring in a pedigree. Specifically,  or 2 according to whether the grand paternal or grand maternal allele is transmitted in the paternal meiosis to the j th offspring.
or 2 according to whether the grand paternal or grand maternal allele is transmitted in the paternal meiosis to the j th offspring.  carries the similar information for the corresponding maternal meiosis, namely, = 3 or 4 according to whether the grand paternal or grand maternal allele was transmitted in the maternal meiosis to the j th offspring.
carries the similar information for the corresponding maternal meiosis, namely, = 3 or 4 according to whether the grand paternal or grand maternal allele was transmitted in the maternal meiosis to the j th offspring.
III. Latent Variable Proportional-odds Logistic Model
This step assesses a potential link between a marker and the trait locus through the inheritance pattern at a locus. We use a proportional-odds logistic model that includes two types of latent random variables to detect association between a marker and a disease locus. The two types of latent variables, and  ,
,  represent: (1) the common genetic or environmental factors in a family that are not observed through the covariates and (2) the genetic susceptibility introduced by the family founders and transmitted to their offspring. Conditional on all of the latent variables and inheritance vectors, within the
represent: (1) the common genetic or environmental factors in a family that are not observed through the covariates and (2) the genetic susceptibility introduced by the family founders and transmitted to their offspring. Conditional on all of the latent variables and inheritance vectors, within the  family, the traits of all nonfounders are independent. Let superscript i denote the family and subscript j denote the
family, the traits of all nonfounders are independent. Let superscript i denote the family and subscript j denote the  nonfounder in the family. Given a trait
nonfounder in the family. Given a trait  taking an ordinal value from
taking an ordinal value from  , the trait of the nonfounder in the family follows the distribution:
, the trait of the nonfounder in the family follows the distribution:

where x is the vector of covariates that is available for each study subject,  is the vector of parameters reflecting the covariate effects on the trait,
is the vector of parameters reflecting the covariate effects on the trait,  is the trait-level-dependent intercept and
is the trait-level-dependent intercept and  indicates the familial and genetic contributions to the trait. The EM algorithm (Dempster et al., 1977) is used to find the maximum-likelihood estimation (MLE) of the parameters. After obtaining the MLEs of the parameters, the log-likelihoods while considering (
indicates the familial and genetic contributions to the trait. The EM algorithm (Dempster et al., 1977) is used to find the maximum-likelihood estimation (MLE) of the parameters. After obtaining the MLEs of the parameters, the log-likelihoods while considering ( ) only and considering both and are computed. The difference between the log-likelihoods is used for determining the significance level of linkage. Under certain regularity conditions, the twice of the difference follows a mixture of chi-square distribution under the null hypothesis (). We have also conducted extensive simulation experiments to derive the distribution of the log-likelihood ratio statistic under the null hypothesis for microsatellite markers and use the simulation result to set the level of suggestive and significance linkage signals.
) only and considering both and are computed. The difference between the log-likelihoods is used for determining the significance level of linkage. Under certain regularity conditions, the twice of the difference follows a mixture of chi-square distribution under the null hypothesis (). We have also conducted extensive simulation experiments to derive the distribution of the log-likelihood ratio statistic under the null hypothesis for microsatellite markers and use the simulation result to set the level of suggestive and significance linkage signals.
IV. Output
This part uses JFreeChart library for GUI.
LOT and GENEHUNTER
LOT and Genehunter (parametric analysis) have equivalent parametrizations when the trait is binary. For clarity, let us assume no residual familial and genetic effects and no covariates (i.e., no
and x). For the parametric analysis in GENEHUNTER, the likelihood at a location t can be written as

where  is the set of all possible inheritance vectors for the i th family, f=(f0, f1, f2) denotes the fixed penetrance parameters that must be specified beforehand,
is the set of all possible inheritance vectors for the i th family, f=(f0, f1, f2) denotes the fixed penetrance parameters that must be specified beforehand,  and
and  is the number of disease allele for the jth individual in the ith family.
is the number of disease allele for the jth individual in the ith family.  corresponds to the disease allele frequency. For any given ,
corresponds to the disease allele frequency. For any given ,  and
and  that control the penetrance of the binary trait in our model as follows
that control the penetrance of the binary trait in our model as follows
 , and thus,
, and thus,
 ,
,  and
and  represent the equivalent parametrization of the penetrance in our model to that in GENEHUNTER.
represent the equivalent parametrization of the penetrance in our model to that in GENEHUNTER.Ascertainment
Sensitivity of Parameter Inputs
Input File Formats
The input file for LOT uses a slightly modified format from supports input files the standard GENEHUNTER (or LINKAGE) format. Two input files are required: a locus data file and pedigree file.
(I) locus file. This file contains information on genetic distances between markers, number of alleles at each locus and their frequencies. We explain this file using the sample.loc.
The first row of five numbers in sample.loc is:
| 31 | 0 | 0 | 5 | 0 |
- 31 is the number of loci; also see LINKAGE manual.
- 0 refers to risk locus; also see LINKAGE manual.
- 0 means not sex linked; also see LINKAGE manual.
- 5 is the designated program code used by LINKAGE, referring to MLINK.
- 0 is the number of covariates. This is an added feature in LOT. If you have two covariates such as sex and age to adjust for, change 0 to 2.
| 0 | 0.0 | 0.0 | 0 |
|
|
- Mutation locus:
= 0, if mutation rates are zero,
= the mutation locus number (input order) for non-zero mutation rates. - Male mutation rate.
- Female mutation rate
- Linkage disequilibrium
0, if loci are assumed to be in linkage equilibrium.
= 1, if loci are in linkage disequilibrium. When loci are in linkage equilibrium, allele frequencies must be given under each locus description; otherwise, haplotype frequencies are provided.
| 1 | 2 | 3 | 4 | 5 | ?? | 31 |
|
|
| 1 | 2 |
- 1 refers to the nature of the trait locus, and always use 1 for LOT
- 2 means that the trait locus is di-allelic
| 0.910000 | 0.090000 |
|
|
| 1 |
|
|
| 0.165000 | 0.575000 | 0.75000 |
|
|
| 3 | 9 | # | D1S468 | ||||
| 0.076492 | 0.014699 | 0.008799 | 0.261774 | 0.055894 | 0.026497 | 0.417558 | 0.132387 |
| 0.005899 | |||||||
|
|
| 3 | 12 | # | D1S1609 | ||||
| 0.005701 | 0.011301 | 0.073407 | 0.240124 | 0.330533 | 0.124312 | 0.096010 | 0.053705 |
| 0.050805 | 0.008501 | 0.002800 | 0.002800 | ||||
|
|
| 0 | 0 |
|
|
- The first 0 means no sex difference
- The second 0 means no interference
| 0.000000 | 12.000000 | 13.710000 | 7.120000 | 8.280000 | 11.410000 | 15.850000 |
| 3.070000 | 13.830000 | 12.530000 | 7.020000 | 4.650000 | 11.820000 | 11.370000 |
| 3.510000 | 11.490000 | 12.210000 | 6.750000 | 4.780000 | 16.430000 | 10.140000 |
| 10.250000 | 6.020000 | 7.700000 | 7.220000 | 6.280000 | 7.570000 | 7.410000 |
| 12.870000 | 7.020000 | |||||
|
|
| 0 |
|
|
| 3 | ||
| 1 | 2 | 3 |
|
|
| Phenotype Y | Level |

|
0 |

|
1 |

|
2 |

|
3 |
| (missing) | 999 |
|
|
(II) Pedigree file. This file must consist of columns with the following information in the correct order (e.g., sample.ped):
Pedigree_ID Person_ID Father_ID Mother_ID Gender Phenotype Marker_genotypes Covariates.
The columns should be separated by spaces or tabs (any number of these is allowed).
- Pedigree ID: pedigree identifier
- Person ID: individual identifier
- Father ID and Mother ID: founders parents are coded as 0. Note: Everyone must have either two parents or no parents in the data set. Enter 0 if one or both parents are not available.
- Gender and, for the gender column (1 = Male and 2 = Female). Note that gender can be re-entered again as duplicated column later to serve as a covariate.
- Phenotype(Y): Missing phenotypes can be coded as 999.
- Marker genotype code: to code a codominant marker locus phenotype, simply list the two numbered alleles with at least one space or tab between the alleles. The unknown genotype is coded as 0 0.
- The covariates such as gender and age.
The pedigree/person IDs are treated as character strings. They do not have to be integers or numbered sequentially. The phenotype and covariates can be integers or reals.
Downloads
Executables
- LOT_Windows.exe
- LOT_Windows_command_line.exe
- LOT_Linux.tar.gz(The javax.swing package is required to be installed in your system before running LOT.)
- LOT_Linux_command_line
- LOT_Mac_command_line
Sample Files
- sample.loc: Sample locus file.
- sample.ped: Sample pedigree file.
- sample.ped.output: Sample output file.
- sample.png;Sample output file.
{kind=link}
Running LOT
a. Running LOT with user-friendly graphical interface on Windows or Linux.
2. Select the input files and click on Add to add them into the project. You can add more than one sets of input files to a project at the same time to have them run sequentially.
Please note that if you are running LOT with the GUI in Linux, please make sure there is no white space in the file names or paths. White spaces in file names and paths will cause a Wrong number of parameters error.
3. Click on Clear if the selected input files need to be discarded and repeat step 2 to select desired input files. Otherwise, click on Run to perform calculations are input files displayed in the File Selected window. Intermediate output produced by the program, which indicates the progress of computation, is displayed in the Intermediate Output window.
4. After computation is done, LOT displays LOT finished and the location of the result file in the Intermediate Output window.
5. You can click on View Result Files to open the dialog that contains the options for displaying the result files. In the dropdown list, select the output file you would like to see.
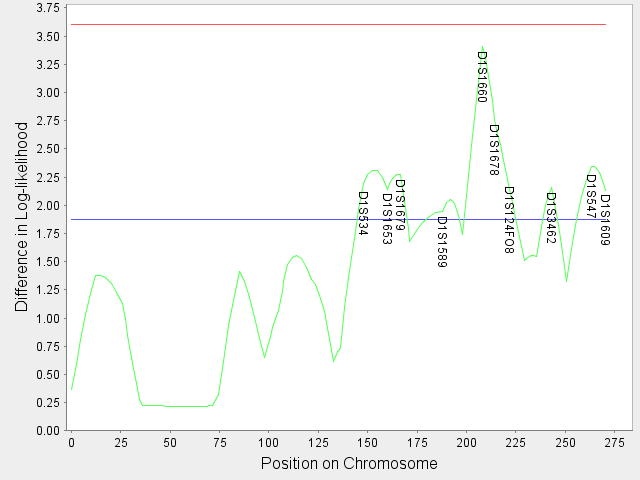
6. Click on View Text and/or View Image to display the tabulated text output and the diagram. In the tabulated text output, the name of each marker, the position of each marker and inter-marker location, the log-likelihood computed without considering any latent variables, the log-likelihood computed with U1 and the log-likelihood computed considering both U1 and U2 are listed. The graphical output plots the difference in log-likelihood while considering both U1 and U2 and just U1 against the positions of the markers and inter-marker locations (the green curve). The blue line and red line are thresholds for suggestive linkage and significant linkage obtained from simulation studies with 400 micro-satellite markers. If the difference in log-likelihood exceeds these thresholds, the name of the markers at that particular location is displayed on the curve.
7. While the tabulated text output is automatically saved into a tab-delimited plain text file, the user has the option to save the diagram in PNG format by selecting Save As in the File menu in the diagram's dialog.
The user also has the option to suppress the marker names displayed on the curve by clicking on the Hide Significant Markers button on the lower left corner of the window.
8. You can save the current project by selecting Save from the File menu of the main program window. By doing so, the next time you open the LOT program you can view the results from this project without repeating the calculation.
9. To open a saved project, click on Open Project in the File menu. Repeat step 5-7 to view the results saved for the project.
b. Running LOT from command line in Windows.
After downloading LOT_Windows_command_line.exe there are two options to evoke it.
- Double click on LOT_Windows_command_line.exe and a DOS window will pop up. The user will be prompted to enter the .ped, .loc and output file names. After the file names are provided, the LOTprogram will start executing.
- In the folder where LOT_Windows_command_line.exe is saved, it can be evoked by typing LOT_Windows_command_line.exe sample.ped sample.loc sample_output.txt in a DOS window. Replace sample.ped, sample.loc and sample_output.txt with the name of the pedigree file, locus file and output file of your choices. If more or less than three file names were provided, the program would prompt the user to enter the file names again.
Please note that if the files are not located in the same folder as the executable, use the full path instead of just the file names. The tab-delimited text output is the only output file provided under this option.
c. Running LOT from command line in Linux.
d. Running LOT from command line in Mac OS X.
Genehunter License Agreement
- Redistributions must reproduce the above copyright notice, this list of conditions and the following disclaimer in the documentation and/or other materials provided with the distribution. Redistributions of source code must also reproduce this information in the source code itself.
- If the program is modified, redistributions must include a notice (in the same places as above) indicating that the redistributed program is not identical to the version distributed by Whitehead Institute.
- All advertising materials mentioning features or use of this software must display the following acknowledgement:
This product includes software developed by the Whitehead Institute for Biomedical Research. - The name of the Whitehead Institute may not be used to endorse or promote products derived from this software without specific prior written permission.
This software is provided by the whitehead institute “as is” and any express or implied warranties, including, but not limited to, the implied warranties of merchantability and fitness for a particular purpose are disclaimed. in no event shall the whitehead institute be liable for any direct, indirect, incidental, special, exemplary, or consequential damages (including, but not limited to, procurement of substitute goods or services; loss of use, data, or profits; or business interruption) however caused and on any theory of liability, whether in contract, strict liability, or tort (including negligence or otherwise) arising in any way out of the use of this software, even if advised of the possibility of such damage.